Population Structure MDS/PCA plot
以ISGC上的SNP50_Breedv2数据为示例数据,已经是PLINK的PED和MAP格式数据。数据和R codes获得方式:</br>
#git shell 下使用如下命令直接在本地建立demo_genetics文件夹或者从相应的地址下载数据
git clone https://github.com/geneticswithme/demo_genetics.gitPLINK_v1.07的--mds-plot和PLINK_v1.09的--pca二选一。
#PLINK_v1.07
plink --sheep --file SNP50_Breedv2 --cluster --mds-plot 10
#生成plink.mds文件是作图文件
#PLINK_v1.09
plink --sheep --file SNP50_Breedv2 --pca
#生成plink.eigenval 和plink.eigenvec文件,eigenvec文件是作图文件用R作图</br>
#首先进入本地的demo_genetics目录下,
#setwd("#path_to_demo_genetics")
setwd("G:/mywork/demo_genetics")
list.files()#查看该目录下的文件,至少要有plink.mds和plink.eigenvec文件
#对plink.mds 文件作图
mds <- read.table("mds.plink", header=T)
head(mds) #查看数据前几行
attach(mds) # 以列名作为变量名将每列数据载入R内存中。
table(FID) #查看家系及数目,这里的家系即是各个品种
plot(C1, C2, col= as.factor(FID)) #以第一mds和第二mds分别为x y坐标做散点图
legend("bottomleft",bty=n, cex=0.8, legend=unique(FID), border=F, fill=unique(as.factor(FID)))
#标注上图例
#以pdf格式保存作图结果
pdf("mdsplot.pdf",width=5,height=5)
plot(C1, C2, col= as.factor(FID))
legend("bottomleft",bty=n, cex=0.8, legend=unique(FID), border=F, fill=unique(as.factor(FID)))
dev.off()
#以png格式保存作图结果
png("mdsplot.png",width=400,height=400)
plot(C1, C2, col= as.factor(FID))
legend("bottomleft",bty=n, cex=0.8, legend=unique(FID), border=F, fill=unique(as.factor(FID)))
dev.off()
detach(mds)#卸载R中mds数据框的列变量名
#对plink.eigenvec文件作图,基本上与上面相同,只需注意文件格式
pca<- read.table("mds.eigenvec", header=F)
colnames(pca)<-c("PopName","Id",paste("PCA",1:20,sep=""))
head(pca)
attach(pca)
table(PopName)
plot(PCA1, PCA2, col= as.factor(PopName)) #以第一mds和第二mds分别为x y坐标做散点图
legend("topright",bty=n, cex=0.8, legend=unique(PopName), border=F, fill=unique(as.factor(PopName)))
#标注上图例
#以pdf格式保存作图结果
pdf("mdsplot.pdf",width=5,height=5)
plot(PCA1, PCA2, col= as.factor(PopName))
legend("topright",bty=n, cex=0.8, legend=unique(PopName), border=F, fill=unique(as.factor(PopName)))
dev.off()
#以png格式保存作图结果
png("mdsplot.png",width=400,height=400)
plot(PCA1, PCA2, col= as.factor(PopName))
legend("topright",bty=n, cex=0.8, legend=unique(PopName), border=F, fill=unique(as.factor(PopName)))
dev.off()
detach(pca)结果如下所示:
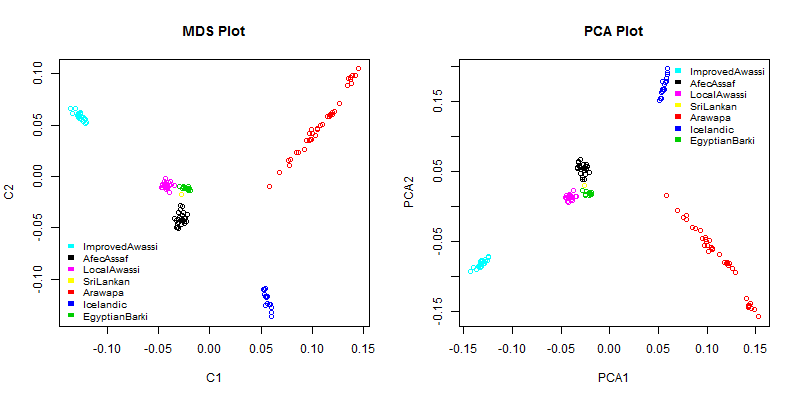 MDS和PCA方法分析群体结构是一样的效果,个体之间都按照所属群体聚成几大类。
MDS和PCA方法分析群体结构是一样的效果,个体之间都按照所属群体聚成几大类。